J. Med. Chem. 2004, 47, 5555-5566 Increased Anti-P-glycoprotein Activity of Baicalein by Alkylation on the A Ring
Yashang Lee,†,| Hosup Yeo,†,‡,| Shwu-Huey Liu,§ Zaoli Jiang,§ Ruben M. Savizky,‡ David J. Austin,‡ andYung-chi Cheng*,†
Department of Pharmacology, Yale University School of Medicine, Department of Chemistry, Yale University, andPhytoCeutica, Inc., New Haven, Connecticut 06520
The aqueous extract of Scutellariae baicalensis Georgi has inhibitory activity against P-gp 170, a multiple drug resistant gene product. Baicalein, one of the major flavones, was found to be responsible for this activity. The hydroxyl groups of the A ring of baicalein were systematically alkylated in order to assess the effect of such modifications on the activity against P-gp 170. The impact of the baicalein modifications on activity against the growth of a human nasopharyngeal cancer cell line KB and its P-gp 170 overexpressing cell line KB/MDR were also examined. The results indicate that alkylation of R5 of baicalein does not have a major impact on the interaction with P-gp 170, whereas alkylation of R6 or R7 alone or both, could enhance the interaction of baicalein with P-gp 170 as well as the amount of intracellular accumulation of vinblastine, a surrogate marker for the activity of P-gp 170 pump of KB/MDR cells. In this case, the optimal linear alkyl functionality is a propyl side chain. These modifications could also alter the activity of compounds inhibiting cell growth. Among the different compounds synthesized, the most potent molecule against P-gp 170 is 5-methoxy- 6,7-dipropyloxyflavone (23). Its inhibitory activity against P-gp 170 is approximately 40 times better, based on EC50 (concentration of the compound enhancing 50% of the intracellular vinblastine accumulation in the KB/MDR cells) and 3 times higher, based on Amax (the intracellular vinblastine accumulation of the KB/MDR cells caused by the compound) as compared to baicalein. Compound 23 is also a more selective inhibitor than baicalein against P-gp 170, because its cytotoxicity is less than that observed for baicalein. The growth inhibitory IC50 of compound 23 against KB and KB/MDR cells are about the same, suggesting that compound 23 is unlikely to be a substrate of P-gp 170 pump. Acetylation of R6, R7 or both could also decrease EC50 and increase Amax. Acetylated compounds are more toxic than baicalein, and their potency against cell growth is compromised by the presence of P-gp 170, suggesting that these compounds are substrates of P-gp 170. Benzylation of R6 or R7 but not both also enhanced anti-P-gp170 activity and potency against cell growth; however, the presence of P-gp 170 in cells did not have an impact on their sensitivity to these molecules, suggesting that the benzylated compounds are inhibitors but not substrates of P-gp 170, and perhaps have a different mechanism of action. In conclusion, the substitutions of R6 and R7 hydroxyl groups by alkoxy groups, acetoxy groups, or benzyloxy groups could yield compounds with different modes of action against P-gp 170 with different mechanisms of action against cell growth. Introduction
blocking absorption by the intestine, excretion of chemi-cals into the bile duct or kidney tubules, prevention of
P-glycoprotein 170 (P-gp 170), a member of the ABC
(ATP Binding Cassette) family, acts as an ATP-depend-
chemicals taken into the brain through the blood-brain
ent drug efflux pump, preventing intracellular ac-
barrier, and efflux of steroid hormones and cholesterol
cumulation of miscellaneous drugs.1,2 Overexpression of
from feces.2,4,5 Developing drugs to inhibit P-gp 170
this protein is one of the mechanisms of multidrug
activity is an important area of drug discovery. Such
resistance (MDR) of cancer cells. This protein is ex-
drugs could have use in facilitating the oral absorption
pressed in a cell- and tissue-specific manner, with high
of drugs through the intestine, or the uptake of chemi-
levels detectable in the kidney, liver, blood-brain
cals that are substrates of P-gp, into the brain. In
barrier, and lining of the intestine.3 Studies using mdr1
addition, these compounds could also potentiate the
knockout mice and P-gp 170 tissue distribution in
action of antitumor drugs, which are substrates of P-gp
humans suggested several physiological roles of P-gp
170 in cancer cells that overexpress the P-gp 170
170, including protection against toxic xenobiotics by
A large number of compounds with major structural
* Corresponding author. Dr. Yung-chi Cheng, Department of Phar-
differences have been found to act as inhibitors or
macology, Yale University School of Medicine, 333 Cedar St., SHM
B254, New Haven, CT 06520. Tel: (203)-785-7119. Fax: (203)-785-7129. E-mail: [email protected].
(VRM), a calcium channel antagonist; trifluoperazine,
† Department of Pharmacology, Yale University School of Medicine.
a calmodulin inhibitor; cyclosporin A (CSA), an immuno-
‡ Department of Chemistry, Yale University. §
suppressant; and progesterone, a steroid hormone.
| The first two authors contributed equally to this manuscript.
Verapamil has been examined clinically in combination
Journal of Medicinal Chemistry, 2004, Vol. 47, No. 22
with cancer chemotherapy.6,7 However, the results were
action. Since baicalein (a 5,6,7-trihydroxyflavone) and
rather unsatisfactory due to high plasma drug levels,
wogonin (a 5,7-dihydroxy-8-methoxyflavone) have a very
required to effectively reverse the MDR phenotype of
similar structure, this raised the possibility of an
cancer cells, which could cause cardiac toxicity. Com-
interesting structure-activity relationship of the fla-
pounds with higher potency against and selectivity for
vone natural products for P-gp 170 inhibition.
P-gp 170 are needed. A second generation of MDR
In this study, we synthesized and evaluated a series
reversal agents has emerged and is based on the
of baicalein analogues, focusing on the substitution
chemical modification of the first generation of inhibi-
pattern at positions 5, 6, and 7 of the A-ring. Our results
tors. Among these, dexniguldipine8 and dexverapamil9
show that alkoxy groups on the A-ring of the flavone
were found to be more selective against P-gp 170, but
greatly increase the anti-P-gp 170 activity and alter
they did not display improved potency. The acridone-
their selectivity for the efflux pump.
carboxamide derivative GF120918 (GG918)10 and thecyclosporin A analogue PSC83311 both displayed an
Chemistry
activity that was 10-30 times more potent than the first
The synthesis of O-substituted baicalein derivatives
generation of modulators, such as verapamil, tamoxifen,
from commercially available reagents was carried out
and cyclosporin A.12 A number of these compounds are
using the general synthetic approach shown in Schemes
currently under clinical evaluation.
1-4. Acetylation of baicalein (1) with acetic anhydride
Flavonoids are important class of natural products
was performed in pyridine to give mono-, di-, and
found in plants. With its polyphenolic structure, this
triacetylated derivatives 2-4. Baicalein was treated
class of compounds has multiple actions, including
with benzyl bromide in dry acetone with potassium
interacting with estrogen receptor, serving as a free-
carbonate to provide mono- and dibenzylated analogues
radical scavenger, and inhibiting protein kinase, NF-
15 and 16. The O-methylated derivatives 6 and 8 of κB and P-gp 170, among others.13 The biological activity
baicalein were readily prepared by reaction with tri-
found in herbal preparations is often attributed to its
methylsilyldiazomethane (TMSCHN2) in a methanolic
flavonoids. For example, the coadministration of grape-
THF solution at room temperature. Since the hydroxyl
fruit juice with various drugs has led to an increase in
function on the C-5 position of baicalein makes an
the plasma concentration of the drugs, which was
intramolecular hydrogen bond with the 4-keto group,
attributed to the bioflavonoids found in the grapefruit
it is resistant to alkylation, and benzylation of baicalein
occurred in the following order: 6 > 7 > 5.
Flavonoids have also been shown to act on multiple
The O-methylated products 6 and 8 of baicalein
targets with different specificity. For example, the
exhibited more potent anti-P-gp activities than that of
flavonoid that binds to estrogen receptor requires hy-
baicalein itself. This finding led us to design and
droxyl groups at positions 2 and 3 of the B-ring, a double
synthesize a series of alkylated baicalein analogues in
bond at positions 2-3 of the C-ring, and the absence of
order to examine their anti-P-gp activity. A variety of
any hydrophobic prenylated substituent.16 This is mark-
alkyl chains were attached to the hydroxyl groups in
edly different than the flavonoids that inhibit various
the baicalein A ring, and related flavonoids, through
ATPases or protein kinases. Recognition of the ATP
phenol alkylation with a number of alkyl halides. The
binding pocket of these proteins requires the presence
reaction of catechols on the baicalein A ring and
of three hydroxyl groups at positions 5 and 7 on the
flavonoids with bromochloromethane in DMF at 50 °C
A-ring and position 3 of the C-ring, which favors some
in the presence of cesium carbonate provided the cor-
flavonols.17 Moreover, some protein kinases exhibit
responding methylenedioxy derivative 19. Similarly,
different structural requirements for binding: an isofla-
compound 13 was readily prepared by heating baicalein
vone structure has been demonstrated to inhibit ty-
at 170 °C for 1 h with dichlorodiphenylmethane and
rosine kinase activity,18 and a flavone substituted at
then reaction with TMSCHN2 in methanolic THF at
position 8 of A-ring with 4-(3-hydroxy-1-methylpiperidi-
nyl) group has demonstrated activity against CDK2.19
The selective brominations at the C-8 position of
The inhibition of P-gp 170 by flavonoids has also been
baicalein and baicalein derivatives were performed
investigated and two binding modes have been postu-
directly with N-bromosuccinimide in the presence of a
lated. The studies were performed using a truncated
catalytic amount of concentrated sulfuric acid at room
form of P-gp 170 in a membrane preparation from P-gp
170 overexpressing cells. The structural requirementsfor flavonoid activity have recently been summarized,20
Biological Results
and it appears that different classes of flavonoids have
In recent studies, we found that the anti-P-gp activity
different structural requirements for inhibitory activity
of Scutellaria baicalensis Georgi could inhibit P-gp 170
and have attributed this activity to the high quantity
In our previous study, we found that Radix scutel-
of baicalein (compound 1) found in the extract. We lariae (Scute), a well-known Chinese herb, has inhibi-
therefore synthesized a number of baicalein-related
tory activity against P-gp 170. The active component of
compounds in order to evaluate their structure-activity
the Scute herb was found to be the natural product
relationship against P-gp 170 activity. Since flavonoids
baicalein. However, the 7-glucuronyl form of baicalein,
could have multiple sites of action that affect cell
which is the most abundant component of Scute, did not
growth, we also evaluated their growth inhibitory
show anti-P-gp 170 activity. Other abundant Scute
activity against KB, a human cancer cell line. If a
components, such as wogonoside and wogonin, were also
compound exhibits cytotoxicity through the inhibition
found to lack inhibitory activity against P-gp 170 efflux
of cell function, in addition to serving as a substrate of
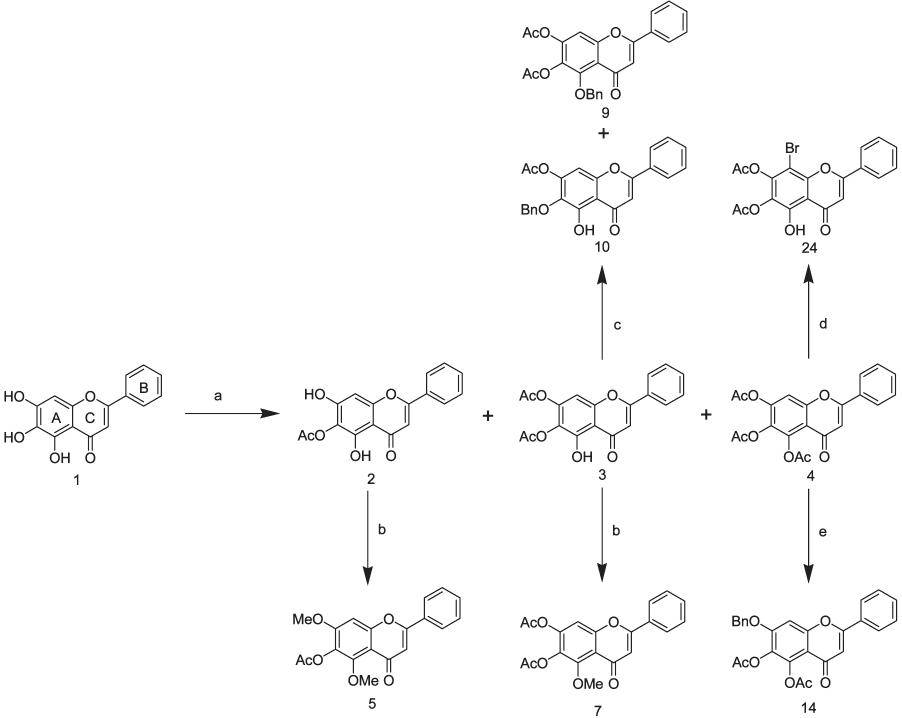
Anti-P-glycoprotein Activity of BaicaleinJournal of Medicinal Chemistry, 2004, Vol. 47, No. 22Scheme 1a a Reagents and conditions: (a) Ac2O, pyridine, rt; (b) TMSCHN2, THF:MeOH (2:1), rt; (c) K2CO3, BnBr, acetone, reflux; (d) NBS, THF,
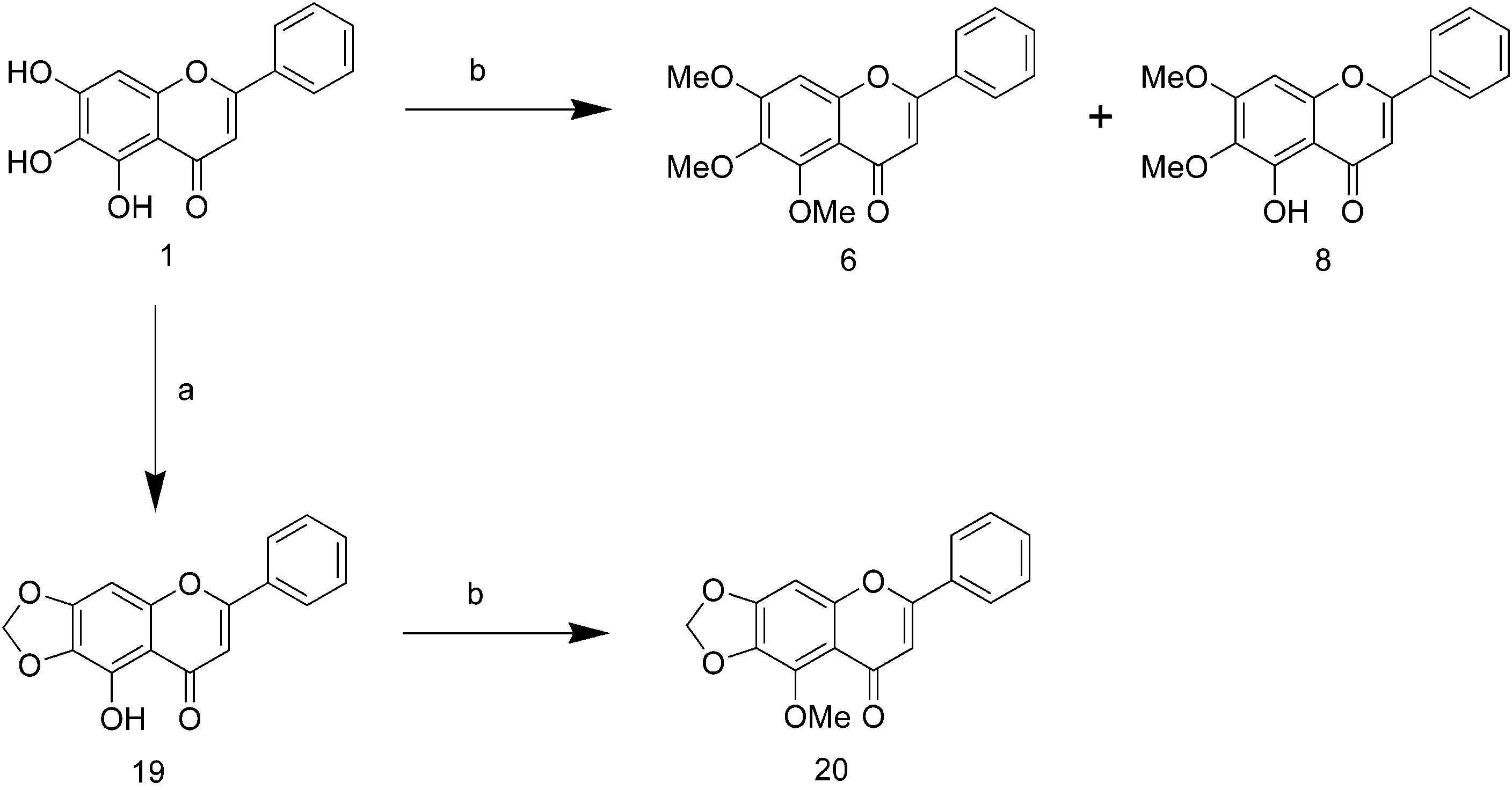
concd H2SO4, rt; (e) K2CO3, KI, BnBr. Scheme 2a a Reagents and conditions: (a) Cs2CO3, BrCH2Cl, DMF, 50 °C; (b) TMSCHN2, THF:MeOH (2:1), rt.
P-gp 170, then the compound would be expected to
5, 6, and 7, since our initial studies indicated that these
demonstrate less activity against a cell line overex-
positions might play a crucial role in P-gp 170 inhibition.
pressing P-gp 170 than the parent cell line. Therefore,
The KB/MDR cells, which overexpress human P-gp
we employed a multi-drug resistant cell line (KB/MDR)
170 protein, were used to evaluate the anti-P-gp 170
in addition to the parent KB cell line, to assess the
activity of drugs. In this manner, the intracellular
susceptibility of a compound to act as a substrate or
amount of vinblastine, a substrate of P-gp 170 pump,
inhibitor of the P-gp 170 efflux pump. Our modification
was measured in the presence of the known P-gp
of the natural products focused primarily on the func-
inhibitors cyclosporin A and verapamil and compared
tional groups of the baicalein A ring, especially positions
to our synthetic flavones. The concentration of com-
Journal of Medicinal Chemistry, 2004, Vol. 47, No. 22Scheme 3a
This suggests that the bromo substitution prevents compound 3 from acting as a substrate of P-gp 170.
By adding one benzyloxy group to the A ring of
baicalein (Table 1), the flavone became a very potent inhibitor of P-gp 170, irrespective of the position or the presence of other functional groups. All the flavones 6,7- diacetoxy-5-benzyloxyflavone (9), 7-acetoxy-6-benzyloxy- 5-hydroxyflavone (10), 5,6-diacetoxy-7-benzyloxyflavone (14), and 6-benzyloxy-5,7-dihydroxyflavone (15) exhib- ited a low EC50 but higher Amax in comparison with baicalein. On the other hand, the benzyloxy group transformed the flavone into a more toxic compound toward both KB and KB/MDR cells than baicalein. This suggests that these compounds have a different site of action in addition to P-gp 170, for which activity is required to maintain cell growth. The fact that these compounds have the same IC50 for KB and KB/MDR cells, suggests that they are inhibitors but not sub- strates of the P-gp 170 efflux pump. When the A ring of baicalein is substituted with two benzyloxy groups, the flavone shows decreased cytotoxicity and anti-P-gp activity, without regard to being cyclic (13) or not (16).
Experimental results for the alkylated baicalein
compounds are shown in Table 2. The introduction of a methoxy group onto the A ring decreased the EC50 and increased the Amax. The presence of two (8) or three (6) a Reagents and conditions: (a) Ac2O, pyridine, rt; (b) TMSCHN2,
methoxy groups on the A ring of baicalein dramatically
THF:MeOH (2:1), rt; (c) K2CO3, BnBr, acetone, reflux; (d) NBS,
increased the anti-P-gp activity, as evidenced by their
EC50 of 4.6 μM and 5.5 μM respectively, without afurther increase in the A
pound required to achieve 50% maximum accumulation
position of the A ring decreased the cytotoxic activity
of vinblastine is presented as the EC50 value. The
maximum accumulation of intracellular vinblastine
max, but did not alter the EC50 against P-gp 170
activity as compared to compounds 8 and 6, regardless
caused by the compounds in 1 h is expressed as Amax
of whether the substitution at R5 is a hydroxy (19) or
(picomoles/106 cells). The cell growth inhibitory activity
methoxy (20) group.
of compounds is presented as the concentration required
Alkyl substitution (methoxy) turned out to be a far
to inhibit 50% growth of KB or KB/MDR cell lines (IC50)
more favorable substitution than either the acetoxy or
following 3 days of compound treatment.
benzyloxy group for flavone anti-P-gp efflux activity.
As shown in Table 1, the number of acetoxy groups
This is most likely due to the fact that this substitution
on the baicalein A ring altered the EC50 of anti-P-gp
prevents the compound from acting as a substrate for
activity. Compounds with one (2) or two (3) acetoxy
P-gp 170 or renders it more specific against P-gp 170.
groups on position 6 and 7 of the A ring exhibit an EC50
For this reason, we further explored the potential of this
that is one-fourth that of the parental compound. The
type of substitution in a search for an optimal linear
activity of the flavone with three acetoxyl groups (4) at
alkoxy group. The ethoxy group was shown to be better
position 5, 6, and 7 did not differ from compound 2 or
than the methoxy group. The EC50 values of 6-ethoxy-
3. The Amax values of these three compounds were also
5,7-dihydroxyflavone (32) and 6,7-diethoxy-5-hydroxy-
similar, but higher than that found for baicalein. The
flavone (33) were 2.3 μM and 1.8 μM, respectively, which
flavones with acetoxy groups were more toxic to KB cells
is about the same as that of CSA. In addition, the ethoxy
than the KB/MDR cells, indicating that the substitu-
compound 33 had a higher Amax and less cytotoxicity
tions of hydroxyl groups by acetoxy groups could render
than either CSA or compound 32. While there is no
the flavone a better substrate for P-gp 170 efflux pump.
significant difference between one and two ethoxy
Substitution of the acetoxy groups in compound 4 with
groups on the EC50, there is a big difference in their
one (7) or two (5) methoxy groups did not alter the EC50
cytotoxicity. Substitution of the R5 hydroxyl group in
or Amax substantially, but increased the IC50 value
compound 33 with methoxy (44) did not change the EC50
against cell growth. We also evaluated the impact of a
significantly. Substitution of the R7 ethoxy group of
bromo group on position 8 on anti-P-gp 170 efflux
compound 33 with a methoxy (38) did not alter the IC50
activity. Compound 25 (8-bromobaicalein) decreased the
EC50 to 15 μM, but the Amax did not change, as compared
Similar to the benzyloxy group substitution, the
to baicalein. Compound 24 (the 8-bromo derivative of
presence of the propyloxy substitution on any position
compound 3) also showed less favorable activity against
of the A ring has a significant effect on the potency and
P-gp 170 activity than compound 3. Both of the com-
degree of anti-P-gp 170 efflux activity. One propyloxy
pounds with an 8-bromo group were toxic to KB and
group on R6 (17) decreases the EC50 to 2 μM and
KB/MDR cells and showed the same IC50 as compound
increases the Amax 9-fold higher than that of the control;
3 to KB cells, but lower than that for the KB/MDR cells.
two propyloxy groups on R6 and R7 (18) decreases the Anti-P-glycoprotein Activity of BaicaleinJournal of Medicinal Chemistry, 2004, Vol. 47, No. 22Scheme 4a a Reagents and conditions: (a) K2CO3, CH3(CH2)nX (X ) I or Br, n ) 1 for 32, 33, n ) 2 for 17, 18, n ) 3 for 45, n ) 4 for 40, n ) 5
for 42, n ) 7 for 34), acetone, reflux; (b) TMSCHN2, THF:MeOH (2:1), rt. Table 1. Anti-P-gp Activity and Cytotoxicity of Modified Baicalein Compounds a clogD was calculated using the following equation: clogD ) clogP - log[1 + 10(pH-pKa)], where clogP is found using ChemDraw ULTRA,
version 6.0.1, for 1-octanol/water system, the pH of the experiment was 7, and the pKa was assumed to be 10 (values ranging from 8 to11 made insignificant changes to the clogD value). b The anti-P-gp efflux activity is represented by intracellular vinblastine accumulationin 1 h with or without drug treatment; see Experimental Section for details. c Maximum vinblastine accumulation (pmoles) of flavones(<100 μM) treated cells in the 106 cells in 1 h. d EC50 calculated as the concentration (μM) that causes 50% of maximum vinblastineaccumulation in the cells in 1 h. e Cytotoxicity IC50 calculated as the concentration (μM) required for 50% inhibition of cell growth, foreach respective cell line, after 72 h of drug exposure. f Control cells were treated with vinblastine only. All values represent the mean (SD of at least three identical experiments.
EC50 to 1.4 μM, and the Amax is 10-fold higher than the
5,6,7-trimethoxyflavone (6), compound 41 to 40 and
control. In addition, the EC50 of 5-hydroxy-7-methoxy-
compound 43 to 42. Finally, the most potent flavone in
6-propyloxyflavone (21) was further decreased to 1.2 μM
this series was found to be 5-methoxy-6,7-dipropyloxy-
and the Amax was 9-fold higher than that of the control.
flavone (23), with two propyloxy groups on R6 and R7
The molecule 5,7-dimethoxy-6-propyloxyflavone (22),
and a methoxy group on R5. The EC50 of compound 23
with an added methoxy on the R5 hydroxyl group,
was found to be 0.9 μM and the Amax is 10-fold higher
showed the same EC50 and Amax as compound 21. The
than that of the control. This is more efficient than CSA,
cytotoxicity of 6,7-diethoxy-5-methoxyflavone (44) was
which shows an activity that is 7-fold higher than
higher than that of 6,7-diethoxy-5-hydroxyflavone (33),
control. The presence of a methoxy group on the R5
which only contains a single R5 position change from
position of compound 23 makes it slightly more toxic
hydroxy to methoxy. The same phenomenon is found
than compound 18 to KB and KB/MDR cells, but this
when comparing 5-hydroxy-6,7-dimethoxyflavone (8) to
cytotoxicity is not altered in the presence of P-gp 170
Journal of Medicinal Chemistry, 2004, Vol. 47, No. 22Table 2. Anti-P-gp Activity and Cytotoxicity of Alkylated Baicalein Compounds a clogD was calculated using the following equation: clogD ) clogP - log[1 + 10(pH-pKa)], where clogP is found using ChemDraw ULTRA,
version 6.0.1, for 1-octanol/water system, the pH of the experiment was 7, and the pKa was assumed to be 10 (values ranging from 8 to11 made insignificant changes to the clogD value). b The anti-P-gp efflux activity is represented by intracellular vinblastine accumulationin 1 h with or without drug treatment; see Experimental Section for details. c Maximum vinblastine accumulation (pmoles) of flavones(<100 μM) treated cells in the 106 cells in 1 h. d EC50 calculated as the concentration (μM) that causes 50% of maximum vinblastineaccumulation in the cells in 1 h. e Cytotoxicity IC50 calculated as the concentration (μM) required for 50% inhibition of cell growth, foreach respective cell line, after 72 h of drug exposure. f Control cells were treated with vinblastine only. All values represent the mean (SD of at least three identical experiments.
activity, which suggests that compound 23 is not a
for binding affinity. The assay used in these studies,
substrate of P-gp 170. Compounds with alkoxy substitu-
however, directly measures the binding affinity of
tions of longer chain length lead to a decrease in Amax,
compounds for the cytosolic nucleotide-binding domain
without altering the EC50 substantially. No obvious
of P-gp 170 protein and does not provide a measure of
correlation between a molecule’s clog D value and anti-
anti-P-gp 170 functional activity in living cells.
Ahcene Boumendjel et al.20 incorporated modified
chrysin in their studies and achieved results similar to
Discussion and Conclusion
our own. Their experiments showed that the increase
Other groups have explored the anti-P-gp activity of
in hydrophobicity of chrysin by alkylation with methyl,
balcalein, which exhibits moderate instability in cell
isopropyl, benzyl, 3,3-dimethylallyl, or geranyl substit-
culture. In drug screens involving molecules isolated
uents correlated with an increase in affinity for in vitro
from Chinese herbal medicines, Thomas Efferth et al.
binding to the P-gp cytosolic domain. Their anti-P-gp
found several compounds exhibiting anti-P-gp activity.21
activity is therefore solely dependent on the number of
However, baicalein was not included in their list. With
isopropyl groups, irrespective of the A-ring position (6,
adjacent trihydroxyl groups on the A ring, baicalein is
7, or 8). Our results indicate that the number of carbons
not a stable compound. It is easily oxidized and forms
permitted in the alkoxy group is limited to three. Alkoxy
a green precipitate within a few hours under assay
groups in all three positions 5, 6, or 7 make a contribu-
conditions. In our 1-h vinblastine accumulation experi-
tion to anti-P-gp activity. Potency is dependent on the
ment, we observed a moderate loss of baicalein. Since
alkoxy(l) group and decreases in the following order:
this molecule is so unstable, it is not surprising that it
propyloxy > ethoxy > methoxy. In our studies, all
was not identified in the original 4-10 day anti-P-gp
flavones with alkoxy groups have the same or better
anti-P-gp activity than that of cyclosporin A (measured
Our previous studies have shown that flavone has the
by vinblastine accumulation) and much higher than that
highest anti-P-gp activity among all of the flavonoid
of the flavones with C-alkyl groups, which only ac-
subclasses, which include flavone, flavonol, isoflavone,
cumulate about 20-30% of drug as compared to cy-
flavanone, and glycosylated flavone. This conclusion is
closporin A. The impact on anti-P-gp activity caused by
consistent with that observed by Gwenaelle Conseil et
n-propyloxy substituents is greater than having C-
al.22 who demonstrated that the 2,3-double bond on C
isopropyl or C-dimethylallyl substituents on these com-
ring and the hydroxyl group on the A ring are important
pounds. Perhaps the high potency observed for their
affinity determinants for flavonoid binding to P-gp. In
best compound, which has an O-dimethylallyl substitu-
addition, Jose M. Perez-victoria et al.23 showed that no
tion pattern, results from the effect of oxygen alkylation
matter how large a substitution is made, its effect is
and not from the dimethylallyl group itself. Moreover,
not as important as the 2,3-double bond in the C ring
the Amax of compound 23 is 167% of cyclosporin A and Anti-P-glycoprotein Activity of BaicaleinJournal of Medicinal Chemistry, 2004, Vol. 47, No. 22
236% of verapamil. One possible interpretation of these
flavones more toxic to cells as compared to the hydroxyl
results is that the alkoxyflavone is more specific for P-gp
170 and has no other cellular effect, which is not the
In the case of a fixed functional group on the R5 and
case of cyclosporin A or verapamil. In this manner, it
R7 position, the molecules can be divided into three
can reach a higher level of apparent anti-P-gp activity
groups, those with two hydroxy groups (compounds 1, 2, 15, 17, and 32), two methoxy groups (compounds 5,
Interestingly, all of the flavones with an alkoxy group
6, and 22), and a hydroxy group on the R5 and methoxy
of 4-8 carbons exhibited the same level of EC50 com-
group on the R7 (compounds 8, 21, and 38). The anti-
pared to compound 23, indicating that maybe the
P-gp activity of the compounds with two hydroxyl groups
binding affinity of this series of alkoxyflavones remain
was largely dependent on the functional group at the
the same, since the long chain substituents did not
R6 position, from high (compounds 1 and 2) to very low
decreased the anti-P-gp activity. One possibility is that
(compounds 15 and 32) EC50. The anti-P-gp efflux
the long carbon chain of the alkoxy group changes the
activity of compounds with two methoxy groups is also
orientation of the flavone (e.g., insertion into plasma
dependent on the functional group at the R6 position,
membrane) relative to the P-gp 170 binding site, so that
showing both medium EC50 (compounds 5 and 6) and
they still bind to P-gp but do not block pump activity.
very low (compound 22) activity. The anti-P-gp 170
The benzyl group substituent also showed a large
activity of compounds with a hydroxy group on R5 and
impact on anti-P-gp activity both in the studies of
a methoxy group on R7 do not show obvious dependence
Ahcene Boumendjel’s20 and ours. The benzyloxy group
on the functional group at R6, since neither the methoxy
has a much stronger effect on the A
ethoxy or propyloxy groups changed the EC
group in both systems. On the other hand, both their
noticeably, however, all three were very good inhibitors.
and our benzyloxy groups are connected to the A-ring
Their inhibitory effectiveness is shows the following
at positions 6 and 7. With the same benzyloxy group,
decreasing order: propyloxy (21) > ethoxy (38) >
the two acetoxy groups of our compound (5,6-diacetoxy-
methoxy (8).
7-benzyloxyflavone, the Amax is 97% as compared to
Compounds with the same functional groups on R5
cyclosporin A) may have some supplementary effect
and R6 could also be divided into four categories. With
compared to their compound (7-benzyloxy-5-hydroxy-
a hydroxy group on R5 and a benzyloxy group on R6
flavone, the Amax is 25% as compared to cyclosporin A).
(compounds 10 and 15), the EC50 values decrease and
The addition of two benzyloxy groups on the A-ring
are not affected by the substituent on R7, except for the
caused the flavone (6, 7-dibenzyloxy-5-hydroxyflavone)
molecule containing two benzyloxy groups (16), which
to lose all of its anti-P-gp activity in our experiments,
lost both anti-P-gp 170 activity and cytotoxicity. The
but (6-benzyl-7-benzyloxy-5-hydroxyflavone) showed an
anti-P-gp 170 activity of compounds with a hydroxy
increase in both binding affinity and drug accumulation
group on R5 and an ethoxy group on R6 was not
in their experiments. The only difference between these
dependent on the functional group at R7 either (com-
two molecules is the benzyl or benzyloxy group on
pounds 32, 38, and 33). The anti-P-gp 170 efflux activity
position 6. It should also be noted that cytotoxicity
of compounds with a hydroxy group on R5 and a
increases greatly with benzyloxy substitution on the
propyloxy group on R6 was also not dependent on the
functional group at the R7 (compounds 17, 21, and 18).
To achieve a more comprehensive overview of the
Even the anti-P-gp activity of the most potent com-
structure-activity relationship of the flavones, we sub-
pounds, containing a methoxy group on R5 and a
categorized them based on the position of their substit-
propyloxy group on R6 was not obviously dependent on
uents. We first organized the same functional group on
the chemical function at R7 (compounds 22 and 23). It
R6 and R7, to see the impact of R5 on anti-P-gp efflux
appears that small substituent changes on position 7
activity. Compounds with the same substitutions on R6
do not have an impact on anti-P-gp activity.
and R7 were divided into four groups: compounds 3, 4,
The inhibition of P-gp 170 efflux activity itself should
7, and 9 with two acetoxy groups on R5 and R7;
not cause cytotoxicity, and our data support this theory
compounds 8 and 6 with two methoxy groups; com-
since there appears to be no correlation between P-gp
pounds 33 and 44 with two ethoxy groups; and com-
inhibition and cytotoxicity in the compounds evaluated
pounds 18 and 23 with two propyloxy groups on the R6
in this study. Therefore, the increased toxicity noted in
and R7 position. The EC50 required to inhibit P-gp 170
some of the molecules is likely due to affinity of the
activity of these four groups decrease in the following
compounds for additional biochemical targets with ATP-
order: two acetoxys > two methoxys > two ethoxys >
binding sites. Several flavonoids have been reported
two propyloxys. In addition, the composition of the R5
to be good inhibitors for a variety of ATP-binding
substituents is not as dominant as the composition of
proteins such as plasma membrane ATPases,24,25 pro-
the same two groups on R6 and R7. The anti-P-gp
tein kinase A,26 protein kinase C,27 serine/threonine
activity of flavones with two acetoxy groups on R6 and
protein kinases,28 tyrosine protein kinase,29 and topo-
R7 is dependent on the functional group in the R5
isomerase II.30 Staurosporine produces high intrinsic
position. The anti-P-gp activity of flavones with two
cytotoxicity in human cells in addition to its anti-P-gp
methoxy or ethoxy groups on R6 and R7 did not vary
activity. On the basis of a structural analysis, R. B.
with a change in the R5 functional group. Flavones with
Wang et al.31 suggested that the isobenzopyrrolidone of
two propyloxy groups on R6 and R7 were the most
staurosporine meets the binding requirement of the
potent inhibitors in this series of compounds, regardless
adenosine moiety of ATP and prevents ATP binding to
of whether R5 is hydroxy or methoxy group. However,
the ATP-binding site. In this comparison, however, the
the methoxy group on the R5 position renders the
A ring of galangin (3, 5, 7-trihydroxyflavone) does not
Journal of Medicinal Chemistry, 2004, Vol. 47, No. 22
overlap well with the five-membered ring of adenosine,
phobic region of P-gp rather than the ATP-binding site.
and that is the reason galangin does not exhibit cyto-
Benzyloxyflavone, on the other hand, may likely be
toxicity as a staurosporine analogue. A. D. Pietro et al.32
overlapping the ATP-binding site, in order to decrease
indicated that among a total of 29 flavonoids examined,
the efflux of P-gp 170 substrates such as vinblastine,
only three flavonols were found to bind to the ATP-
and be exhibiting a high intrinsic cytotoxicity through
binding site, and the hydroxyl group at position 3 of the
interaction with the ATP-binding sites of other vital
flavonol is critical for binding. This requirement is
similar to that observed for quercetin binding to the Hck
In conclusion, acetylation, alkylation, or benzylation
tyrosine kinase, as demonstrated by cocrystallization33
of hydroxyl groups on the A ring of baicalein can
and for other ATPases by inhibition kinetics.34 On the
enhance interaction with the P-gp 170 protein and
basis of these observations, most of our synthetic
prevent its substrate efflux activity. The mode of
flavones are unlikely to be good candidates for ATP-
interaction with these modified flavones appear be
binding site affinity because they lacking the hydroxyl
quite different, given the broad differences noted in
group on R3, except the benzyloxy flavone which could
EC50, Amax and cytotoxicity. The alkoxyflavones may
potentially block ATP binding by fitting to the adenine
interact with P-gp at a site other than the ATP-binding
site in the ATP-binding pocket like staurosporine.
site, whereas the other modified flavonoids likely mimic
When a benzyloxy group is connected to the A-ring of
the adenosine moiety of ATP and block the ATP-binding
baicalein, its structure resembles L868276 (5,7-dihydroxy-
site. This suggests that the alkoxyflavones may have a
8-[4-(3-hydroxy-1-methyl)piperidinyl]-flavone), which fits
lower propensity to interact with the ATP-binding site
nicely to the adenine-binding pocket of CDK2,19 and this
of other proteins, as observed by their lower cytotoxicity.
may be a source of its increased cytotoxicity. Interest-
In summary, the alkoxyflavones appear to be quite
ingly, the cytotoxicity is lost when we add another
promising modulators of P-gp 170 function and warrant
benzyloxy group to the A-ring of baicalein. This is
similar to a pattern noted with staurosporine modifica-tion: the addition of one benzyl group causes a decrease
Experimental Section
in cytotoxicity (CGP41251),35 and adding another totally
General Chemistry Methods. All solvents and reagents
abolishes the cytotoxic effect (CGP42700).36 It is possible
were obtained from commercial suppliers and were used
that the increased steric bulk added to the molecule by
without further purification. Unless otherwise specified, reac-
virtue of the second benzyl group renders the com-
tions were performed under a nitrogen atmosphere withexclusion of moisture. All reaction mixtures were magnetically
pounds incapable of entering the adenine-binding pocket
stirred and monitored by TLC using Si250F precoated plates
of CDK2 due to steric hindrance. We are currently
from J. T. Baker (0.25 mm). Flash column chromatography
was performed on 32-63 D 60 Å silica gel from ICN SiliTech
The nucleotide binding site of P-glycoprotein contains
(ICN Biomedicals GmbH). Melting points were determined
a region that interacts with hydrophobic steroid deriva-
with an Electrothermal capillary melting point apparatus andare uncorrected. 1H NMR spectra were recorded on a Bruker
tives, such as RU486, called the steroid binding hydro-
AM-400, Bruker AM-500, or GE QE-plus 300 spectrometer.
phobic region (SBHR). This region is most likely located
Chemical shifts are reported using chloroform-d (δ 7.24 ppm)
in close proximity to the ATP binding site since RU486
or DMSO-d6 (2.50 ppm). All coupling constants are described
completely prevents or displaces the hydrophobic nucle-
in Hz. Mass spectra were conducted at the Mass Spectrometry
otide derivative, 2(3)-methylanthraniloyl-ATP (MANT-
Laboratory of the University of Illinois.
ATP).37 A tentative model for the interaction of fla-
6-Acetoxy-5,7-dihydroxyflavone (2). Baicalein 1 (54 mg,
vonoids with P-glycoprotein and related multidrug
0.2 mmol) was dissolved in acetic anhydride (1 mL) andpyridine (1 mL), and the solution was stirred at room tem-
transporters has been proposed by A. D. Pietro et al.32
perature for 2 h. The reaction mixture was poured into ice-
Galangin (5,7-dihydroxyflavonol), kaempferol (4′,5,7-
water (10 mL), and the precipitate was collected by filtration
trihydroxyflavone), kaempferide (3,5,7-trihydroxy-4′-
and purified by flash chromatography on a column of silica
methoxyflavone), and dehydrosilybin (3, 5, 7-trihydroxy-
gel eluted with CH2Cl2/MeOH (20:1) to yield compound 2 (35
3′-monolignolflavone) appear to interact with the cytosolic
mg, 56%) as a yellow powder. mp 205-207 °C; 1H NMR
nucleoside binding domain; the hydroxyl groups at
(DMSO-d6) δ 2.28 (s, 3H), 6.67 (s, 1H), 7.01 (s, 1H), 7.57 (m,3H), 8.79 (m, 2H), 11.28 (s, 1H), 12.97 (s, 1H,); MS (EI) m/z
positions 3 and 5, in addition to the ketone at position
4, are proposed to bind the ATP-binding site, whereas
6,7-Diacetoxy-5-hydroxyflavone (3) and 5,6,7-Triac-
other parts of the molecules bind in the vicinal SBHR
etoxyflavone (4). Baicalein 1 (216 mg, 0.8 mmol) was
region. Prenylation of the A-ring would therefore in-
dissolved in acetic anhydride (40 mL) and pyridine (12 mL),
crease the hydrophobic interactions with both the cy-
and the solution was stirred at room temperature for 48 h.
tosolic steroid-interacting region and the drug-binding
The reaction mixture was poured into ice-water (100 mL), and
site. This would potentially produce a significant shift
the precipitate was collected by filtration and purified by flashchromatography on a column of silica gel eluted with CH
in flavonoid positioning, in such a way that overlap with
MeOH (40:1) to give compounds 3 (72 mg, 25%) and 4 (220
the ATP binding site can no longer occur. Such a prenyl-
mg, 69%) as a pale yellow powder, respectively. (3): mp 204-
flavonoid positioning appears to be efficient enough to
206 °C; 1H NMR (CDCl3) δ 2.36 (s, 6H), 6.74 (s, 1H), 6.98 (s,
directly inhibit P-gp 170 substrate binding and trans-
1H), 7.55 (m, 3H), 7.89 (m, 2H), 12.95 (s, 1H); MS (EI) m/z
port, while indirectly interfering with ATP hydrolysis
354 [M]+, 312, 270 (base). (4): mp 194-195 °C; 1H NMR
and energy transduction. In our case, flavones without
(CDCl3) δ 2.36, 2.37, 2.46 (each s, 9H), 6.67 (s, 1H), 7.52 (s,1H), 7.54 (m, 3H), 7.87 (m, 2H); MS (EI) m/z 396 [M]+, 354,
the 3-hydroxyl group may be acting in a similar fashion,
since the alkoxyl group helps trihydroxyflavone (baica-
6-Acetoxy-5,7-dimethoxyflavone (5). To a stirred solution
lein) to be a better P-gp modulator with less cytotoxicity
of 2 (25 mg, 0.08 mmol) in a mixture of MeOH (4 mL) and
and may be interacting with the steroid binding hydro-
THF (8 mL) was added trimethylsilyldiazomethane (TM-
Anti-P-glycoprotein Activity of BaicaleinJournal of Medicinal Chemistry, 2004, Vol. 47, No. 22
SCHN2, 2 M in hexanes, 0.4 mL, 0.8 mmol). The reaction
and anhydrous K2CO3 (30 mg) in acetone (15 mL) was refluxed
mixture was stirred at room temperature for 12 h and then
for 24 h with stirring. The reaction mixture was filtered, and
evaporated. Flash chromatography of the residue, eluting with
the solvent was evaporated under reduced pressure. Flash
n-hexane/EtOAc (1:1), gave compound 5 (5 mg, 18%) as a pale
chromatography of the residue, eluting with n-hexane/EtOAc
yellow powder. mp 208-210 °C; 1H NMR (CDCl3) δ 2.40 (s,
(3:2), afforded compound 14 (20 mg, 52%) as a white solid. mp
3H), 3.96 (s, 6H), 6.70 (s, 1H), 6.88 (s, 1H), 7.54 (m, 3H), 7.88
174-175 °C; 1H NMR (CDCl3) δ 2.27, 2.48 (s, 6H), 5.14 (s, 2H),
(m, 2H); MS (EI) m/z 340 [M]+, 298, 280 (base).
6.53 (s, 1H), 6.97 (s, 1H), 7.38 (m, 5H), 7.47 (m, 3H), 7.76 (m,
5,6,7-Trimethoxyflavone (6). To a stirred solution of 1 (54
2H); MS (EI) m/z 444 [M]+, 402, 360, 269 (base).
mg, 0.2 mmol) in a mixture of MeOH (6 mL) and THF (12 mL)
6-(Benzyloxy)-5,7-dihydroxyflavone (15) and 6,7-(Diben-
was added TMSCHN2 (2 M in hexanes, 1.2 mL, 2.4 mmol). zyloxy)-5-hydroxy-flavone (16). A mixture of 1 (54 mg, 0.2
The reaction mixture was stirred at room temperature for 36
mmol), benzyl bromide (0.12 mL), and anhydrous K2CO3 (83
h and evaporated. Flash chromatography of the residue,
mg) in acetone (15 mL) was refluxed for 8 h with stirring. The
eluting with CH2Cl2/acetone (15:1), gave compound 6 (30 mg,
reaction mixture was filtered, and the solvent was evaporated
48%) as a pale yellow powder. mp 164-165 °C; 1H NMR
under reduced pressure. Flash chromatography of the residue,
(CDCl3) δ 3.90, 3.97, 3.99 (s, 9H), 6.67 (s, 1H), 6.81 (s, 1H),
eluting with CH2Cl2/MeOH (100:1 to 50:1), afforded compound
7.50 (m, 3H,), 7.86 (m, 2H); 13C NMR (CDCl3) δ 56.71, 61.96,
15 (30 mg, 42%) as a yellow powder and compound 16 (24 mg,
62.60, 96.67, 108.84, 126.39, 129.38, 131.68, 132.03, 154.98,
27%) as a pale yellow powder. (15): mp 195-197 °C; 1H NMR
158.20; MS (EI) m/z 312 [M]+, 297 (base). HRMS for C18H16O5
(CDCl3) δ 5.27 (s, 2H), 6.67 (s, 2H), 7.44 (m, 5H), 7.52 (m, 3H),
[M]+: calculated, 312.0998; found, 312.0995.
7.88 (m, 2H); 13C NMR (CDCl3) δ 71.78, 92.25, 105.87, 106.69,
6,7-Diacetoxy-5-methoxyflavone (7). To a stirred solution
126.69, 127.99, 129.02, 129.28, 129.48, 130.39, 131.86, 132.18,
of 3 (25 mg, 0.07 mmol) in a mixture of MeOH (2 mL) and
135.77, 146.35, 150.93, 152.22, 164.57, 183.09; MS(EI) m/z 360
THF (4 mL) was added TMSCHN2 (2 M in hexanes, 0.21 mL,
[M]+, 269 (base). HRMS for C22H16O5 [M]+: calculated, 360.0998;
0.42 mmol). The reaction mixture was stirred at room tem-
found, 360.0996. (16): mp 191-193 °C; 1H NMR (CDCl3) δ
perature for 12 h and evaporated. Flash chromatography of
5.17, 5.19 (s, 4H), 6.59 (s, 1H), 6.69 (s, 1H), 7.30-7.55 (m, 13H),
the residue, eluting with CH2Cl2/acetone (30:1), gave compound
7.87 (m, 2H); 13C NMR (CDCl3) δ 71.36, 75.25, 92.38, 106.07,
7 (6 mg, 23%) as a pale yellow powder. mp 240-242 °C; 1H
126.68, 127.68, 128.36, 128.60, 128.68, 129.05, 120.10, 129.49,
NMR (CDCl3) δ 2.35, 2.45 (s, 6H), 3.95 (s, 3H), 6.61 (s, 1H),
131.76, 132.22, 136.16, 137.89, 153.64, 153.94, 158.59, 164.38,
6.96 (s, 1H), 7.52 (m, 3H), 7.85 (m, 2H); MS (EI) m/z 368 [M]+,
183.16; MS (EI) m/z 450 [M]+, 359, 269, 91 (base). HRMS for
C29H22O5 [M]+: calculated, 450.1467; found, 450.1465. 5-Hydroxy-6,7-dimethoxyflavone (8). To a stirred solu- 5,7-Dihydroxy-6-propyloxyflavone (17) and 5-Hydroxy-
tion of 1 (54 mg, 0.2 mmol) in a mixture of MeOH (6 mL) and 6,7-dipropyloxyflavone (18). A mixture of 1 (54 mg, 0.2
THF (12 mL) was added TMSCHN2 (2 M in hexanes, 0.6 mL,
mmol), n-propyl iodide (0.06 mL), and anhydrous K2CO3 (110
1.2 mmol). The reaction mixture was stirred at room temper-
mg) in acetone (20 mL) was refluxed with stirring for 24 h.
ature for 8 h and evaporated. Flash chromatography of the
The reaction mixture was concentrated under reduced pres-
residue, eluting with CH2Cl2/acetone (40:1 to 20:1), gave
sure, diluted with water (30 mL), and extracted with CH2Cl2
compound 8 (8 mg, 13%) as a pale yellow powder. mp 159-
(3 × 30 mL). The extract was washed with water and dried
160 °C; 1H NMR (CDCl3) δ 3.94, 3.99 (s, 6H), 6.59 (s, 1H), 6.70
over MgSO4, and the solvent was evaporated in vacuo. The
(s, 1H), 7.55 (m, 3H), 7.89 (m, 2H); 13C NMR (CDCl3) δ 56.78,
residue was purified by flash chromatography on a column of
61.31, 91.04, 106.07, 126.68, 129.53, 131.73, 132.27, 153.46,
silica gel eluted with CH2Cl2/MeOH (70:1 to 50:1) to yield
159.31, 164.37, 183.16; MS (EI) m/z 298 ([M]+, base), 283.
compounds 17 (7 mg, 11%) and 18 (44 mg, 62%) as a yellow
HRMS for C17H14O5 [M]+: calculated, 298.0841; found, 298.0844.
powder, respectively. (17): mp 162-163 °C; 1H NMR (CDCl3) 6,7-Diacetoxy-5-(benzyloxy)flavone (9) and 7-Acetoxy- δ 1.11 (t, 3H, J ) 7.5 Hz), 1.95 (sextet, 2H, J ) 7.5 Hz), 4.13
6-(benzyloxy)-5-hydroxyflavone (10). A mixture of 3 (21
(t, 2H, J ) 7.5 Hz), 6.62 (s, 1H), 6.69 (s, 1H), 7.54 (m, 3H),
mg, 0.06 mmol), benzyl bromide (0.03 mL), and anhydrous K
107.99, 108.54, 128.79, 131.62, 132.22, 134.04, 134.29, 148.21,
3 (26 mg) in acetone (15 mL) was refluxed for 8 h with
stirring. The reaction mixture was filtered, and the solvent
153.22, 154.79, 165.56, 185.24; MS (EI) m/z 312 [M]+, 297, 283,
was evaporated under reduced pressure. Flash chromatogra-
270 (base). HRMS for C18H16O5 [M]+: calculated, 312.0998;
phy of the residue, eluting with n-hexane/EtOAc (3:2), afforded
found, 312.0993. (18): mp 89-91 °C; 1H NMR (CDCl3) δ 1.07,
compounds 9 (18 mg, 68%) and 10 (8 mg, 33%) as a pale yellow
1.11 (t, 6H, J ) 7.5 Hz), 1.82, 1.92 (sextet, 4H, J ) 7.5 Hz),
powder, respectively. (9): mp 175-177 °C; 1H NMR (CDCl
4.02, 4.05 (t, 4H, J ) 7.5 Hz), 6.54 (s, 1H), 6.66 (s, 1H), 7.53
δ 2.32, 2.47 (s, 6H), 5.22 (s, 2H), 6.61 (s, 1H), 7.02 (s, 1H),
(m, 3H), 7.88 (m, 2H); 13C NMR (CDCl3) δ 10.86, 10.92, 22.78,
7.42 (m, 5H), 7.52 (m, 3H), 7.84 (m, 2H); MS (EI) m/z 444 [M]+,
23.81, 30.09, 71.06, 75.43, 91.67, 106.02, 106.54, 126.64,
402, 360, 269 (base). (10): mp 184-185 °C; 1H NMR (CDCl
129.47, 131.87, 132.13, 132.47, 153.63, 153.71, 159.22, 164.19,
δ 2.37 (s, 3H), 5.22 (s, 2H), 6.64 (s, 1H), 6.70 (s, 1H), 7.42 (m,
183.13; MS (EI) m/z 354 ([M]+, base), 325, 311, 283, 270. HRMS
5H), 7.55 (m, 3H), 7.88 (m, 2H), 13.00 (s, 1H); MS (EI) m/z
for C21H22O5 [M]+: calculated, 354.1467; found, 354.1464. 5-Hydroxy-6,7-(methylenedioxy)flavone (19). A mixture 6,7-(Diphenylmethylenedioxy)-5-methoxyflavone (13).
of 1 (81 mg, 0.3 mmol) and cesium carbonate (244 mg, 0.75
A mixture of 1 (27 mg, 0.1 mmol) and dichlorodiphenylmethane
mmol) in DMF (5 mL) was stirred at room temperature for 30
(0.02 mL, 0.1 mmol) was stirred under nitrogen at 170 °C for
min. Bromochloromethane (0.05 mL, 0.75 mmol) was added
1 h. The reaction mixture was cooled to 30 °C and then
to the DMF solution, and the mixture was stirred at 50 °C for
dissolved in a minimum amount of CH2Cl2. The crude product
8 h then diluted with CH2Cl2. The dichloromethane solution
was purified by flash chromatography on a column of silica
was washed with water and brine, dried over MgSO4, filtered,
gel eluted with CH2Cl2 to yield compound 11 (35 mg, 81%).
and concentrated in vacuo. The residue was purified by flash
To a stirred solution of 11 (14 mg, 0.03 mmol) in a mixture of
chromatography on a column of silica gel eluted with CH2Cl2/
MeOH (2 mL) and THF (4 mL) was added TMSCHN2 (2 M in
MeOH (100:1 to 50:1) to give compound 19 (29 mg, 33%) as a
hexanes, 0.1 mL, 0.2 mmol). The reaction mixture was stirred
pale yellow powder. mp 213-215 °C; 1H NMR (CDCl3) δ 6.12
at room temperature for 24 h and then evaporated. Flash
(s, 2H), 6.61 (s, 1H), 6.70 (s, 1H), 7.55 (m, 3H), 7.87 (m, 2H);
chromatography of the residue, eluting with CH2Cl2/MeOH (40:
MS (EI) m/z 282 ([M]+, base), 149.
1), gave compound 13 (13 mg, 90%) as a pale yellow powder. 5-Methoxy-6,7-(methylenedioxy)flavone (20). To a stirred
mp 238-240 °C; 1H NMR (CDCl3) δ 4.24 (s, 3H), 6.67 (s, 1H),
solution of 19 (17 mg, 0.06 mmol) in a mixture of MeOH (3
6.81 (s, 1H), 7.42 (m, 6H), 7.50 (m, 3H), 7.61 (m, 4H), 7.85 (m,
mL) and THF (6 mL) was added TMSCHN2 (2 M in hexanes,
2H); MS (EI) m/z 448 [M]+, 402, 371, 266, 167 (base).
0.3 mL, 0.6 mmol). The reaction mixture was stirred at room
5,6-Diacetoxy-7-(benzyloxy)flavone (14). A mixture of
temperature for 24 h and evaporated. Flash chromatography
4 (34 mg, 0.086 mmol), benzyl bromide (0.05 mL), KI (3.5 mg),
of the residue, eluting with CH2Cl2/acetone (30:1), afforded
Journal of Medicinal Chemistry, 2004, Vol. 47, No. 22
compound 20 (12 mg, 68%) as a white solid. mp 202-204 °C;
34%) and 33 (12 mg, 12%) as a yellow powder, respectively.
1H NMR (CDCl3) δ 4.14 (s, 3H), 6.08 (s, 2H), 6.71 (s, 1H), 6.75
(32): mp 190-192 °C; 1H NMR (CDCl3) δ 1.55 (t, 3H, J ) 6.9
(s, 1H), 7.51 (m, 3H), 7.86 (m, 2H); MS (EI) m/z 296 [M]+, 268
Hz), 4.23 (q, 2H, J ) 6.9 Hz), 6.60 (s, 1H), 6.68 (s, 1H), 7.53
(m, 3H), 7.89 (m, 2H), 12.50 (s, 1H); 13C NMR (CDCl3) δ 14.04,
5-Hydroxy-7-methoxy-6-propyloxyflavone (21) and 5,7-
64.57, 90.44, 104.87, 105.42, 125.67, 128.50, 129.05, 130.91,
Dimethoxy-6-propyloxy-flavone (22). To a stirred solution
131.17, 145.08, 150.12, 151.56, 163.46, 182.10; MS (EI) m/z
of 17 (19 mg, 0.06 mmol) in a mixture of MeOH (4 mL) and
298 ([M]+, base), 283, 270, 269, 254. HRMS for C17H14O5 [M -
H]+: calculated, 297.0763; found, 297.0762. (33): mp 132-
0.4 mmol). The reaction mixture was stirred at room temper-
133 °C; 1H NMR (CDCl3) δ 1.41, 1.53 (t, 6H, J ) 7.2 Hz), 4.13,
ature for 24 h and evaporated. Flash chromatography of the
4.19 (q, 4H, J ) 7.2 Hz), 6.55 (s, 1H), 6.67 (s, 1H), 7.54 (m,
3H), 7.89 (m, 2H,), 12.65 (s, 1H); 13C NMR (CDCl
2Cl2/MeOH (30:1), gave compound 21
(4 mg, 20%) as a yellow powder and compound 22 (9.2 mg,
15.94, 65.18, 69.30, 91.65, 106.03, 106.55, 126.66, 129.50,
45%) as a pale yellow powder. (21): mp 112-113 °C; 1H NMR
131.83, 132.08, 132.19, 153.69, 153.79, 159.08, 164.24, 183.13;
MS (EI) m/z 326 [M]+, 311, 297 (base), 269. HRMS for C
3) δ 1.12 (t, 3H, J ) 7.5 Hz), 1.95 (sextet, 2H, J ) 7.5
Hz), 3.93 (s, 3H), 4.09 (t, 2H, J ) 7.5 Hz), 6.57 (s, 1H), 6.69 (s,
[M]+: calculated, 326.1154; found, 326.1155.
1H), 7.55 (m, 3H), 7.90 (m, 2H); 13C NMR (CDCl3) δ 10.91,
5-Hydroxy-6,7-(dioctyloxy)flavone (34). A mixture of 1
22.73, 61.23, 71.13, 91.77, 106.02, 106.54, 126.66, 129.49,
(81 mg, 0.3 mmol), 1-iodooctane (0.16 mL), and anhydrous K2-
131.81, 132.19, 133.30, 153.55, 153.73, 159.00, 164.28, 183.11;
CO3 (166 mg) in acetone (25 mL) was refluxed with stirring
MS (EI) m/z 326 ([M]+, base), 283, 269. HRMS for C19H18O5
for 30 h. The reaction mixture was concentrated under reduced
[M]+: calculated, 326.1154; found, 326.1149. (22): mp 135-
pressure, diluted with water (50 mL), and extracted with CH2-
136 °C; 1H NMR (CDCl3) δ 1.13 (t, 3H, J ) 7.5 Hz), 1.96 (sextet,
Cl2 (50 mL × 3). The extract was washed with water and dried
2H, J ) 7.5 Hz), 3.92, 4.01 (s, 6H), 4.09 (t, 2H, J ) 7.5 Hz),
over MgSO4, and the solvent was evaporated in vacuo. The
6.73 (s, 1H), 6.82 (s, 1H), 7.52 (m, 3H), 7.89 (m, 2H); 13C NMR
residue was purified by flash chromatography on a column of
(CDCl3) δ 10.93, 22.71, 58.92, 61.86, 62.63, 71.10, 97.28, 108.62,
silica gel and eluted with CH2Cl2/MeOH (100:1 to 50:1) to give
113.62, 115.71, 121.02, 126.43, 129.38, 130.02, 131.72, 132.00,
compound 34 (122 mg, 82%) as a pale yellow powder. mp 85-
141.00, 153.05, 155.02, 157.98, 161.66, 177.67; MS (EI) m/z
86 °C; 1H NMR (CDCl3) δ 0.89, 0.90 (t, 6H, J ) 6.9 Hz), 1.31-
340 [M]+, 325 (base), 283. HRMS for C20H20O5 [M]+: calcu-
1.52 (m, 20H), 1.79, 1.89 (t, 4H, J ) 6.9 Hz), 4.03, 4.07 (t, 4H,
J ) 6.9 Hz), 6.53 (s, 1H), 6.64 (s, 1H), 7.51 (m, 3H), 7.86 (m,
5-Methoxy-6,7-dipropyloxyflavone (23). To a stirred
2H), 12.45 (s, 1H); MS (EI) m/z 494 [M]+, 382, 270 (base).
solution of 18 (28 mg, 0.08 mmol) in a mixture of MeOH (4 6-Ethoxy-5-hydroxy-7-methoxyflavone (38). To a stirred
mL) and THF (8 mL) was added TMSCHN2 (2 M in hexanes,
solution of 32 (10 mg, 0.034 mmol) in a mixture of MeOH (3
0.32 mL, 0.64 mmol). The reaction mixture was stirred at room
mL) and THF (6 mL) was added TMSCHN2 (2 M in hexanes,
temperature for 24 h and evaporated. Flash chromatography
0.02 mL, 0.04 mmol). The reaction mixture was stirred at room
of the residue, eluting with CH2Cl2/acetone (15:1), gave
temperature for 24 h and evaporated. Flash chromatography
compound 23 (23 mg, 78%) as a pale yellow powder. mp 109-
of the residue, eluting with CH2Cl2/MeOH (50:1), afforded
110 °C; 1H NMR (CDCl3) δ 1.08, 1.12 (t, 6H, J ) 7.5 Hz), 1.82,
compound 38 (7.4 mg, 70%) as a pale yellow powder. mp 133-
1.95 (sextet, 4H, J ) 7.5 Hz), 3.99 (s, 3H), 4.00, 4.06 (t, 4H, J
134 °C; 1H NMR (CDCl3) δ 1.55 (t, 3H, J ) 6.9 Hz), 3.93 (s,
) 7.5 Hz), 6.72 (s, 1H), 6.81 (s, 1H), 7.52 (m, 3H), 7.89 (m,
3H), 4.21 (q, 2H, J ) 6.9 Hz), 6.57 (s, 1H), 6.69 (s, 1H), 7.56
2H); 13C NMR (CDCl3) δ 10.95, 22.76, 23.89, 62.46, 71.00,
(m, 3H), 7.90 (m, 2H), 12.68 (s, 1H); 13C NMR (CDCl3) δ 15.01,
76.28, 97.19, 108.75, 113.16, 126.38, 129.36, 131.61, 132.11,
61.19, 65.25, 91.72, 106.04, 106.58, 126.66, 129.49, 131.81,
140.26, 153.17, 154.90, 158.13, 161.47, 177.69.; MS (EI) m/z
132.19, 133.29, 153.55, 153.72, 158.75, 164.30, 183.11; MS (EI)
368 [M]+, 325 (base), 283. HRMS for C22H24O5 [M]+: calcu-
m/z 312 ([M]+, base), 283. HRMS for C18H16O5 [M]+: calculated,
6,7-Diacetoxy-8-bromo-5-hydroxyflavone (24). A mix- 5-Hydroxy-6,7-(dipentyloxy)flavone (40). A mixture of
ture of 4 (84 mg, 0.21 mmol) and N-bromosuccinimide (NBS, 1 (51 mg, 0.19 mmol), 1-iodopentane (0.076 mL), and anhy-
56 mg, 0.32 mmol) in THF (8 mL) and concd H2SO4 (10 μL)
drous K2CO3 (110 mg) in acetone (20 mL) was refluxed with
was stirred at room temperature for 48 h. The reaction mixture
stirring for 24 h. The reaction mixture was concentrated under
was extracted with EtOAc, washed with 10% aqueous NaHSO4
reduced pressure, diluted with water (40 mL), and extracted
solution and water, dried over MgSO4, and then concentrated
with CH2Cl2 (40 mL × 3). The extract was washed with water
in vacuo. The residue was recrystallized from MeOH to give
and dried over MgSO4 and the solvent evaporated in vacuo.
compound 24 (50 mg, 55%) as a yellow powder. mp 244-246
The residue was purified by flash chromatography on a column
°C; 1H NMR (CDCl3) δ 2.38, 2.44 (each s, 6H), 6.82 (s, 1H),
of silica gel and eluted with CH2Cl2/MeOH (60:1) to give
7.59 (m, 3H), 8.01 (m, 2H); MS (EI) m/z 434 [M + 2]+, 432
compound 40 (68 mg, 87%) as a pale yellow powder. mp 111-
112 °C; 1H NMR (CDCl3) δ 0.94, 0.97 (t, 6H, J ) 6.6 Hz), 1.39-
8-Bromo-5,6,7-trihydroxyflavone (25). A mixture of 1 (35
1.52 (m, 8H), 1.81, 1.91 (quintet, 4H, J ) 6.6 Hz), 4.05, 4.09
mg, 0.13 mmol) and NBS (33 mg, 0.19 mmol) in THF (4 mL)
(t, 4H, J ) 6.6 Hz), 6.56 (s, 1H), 6.68 (s, 1H), 7.54 (m, 3H),
7.90 (m, 2H); MS (EI) m/z 410 [M]+.
2SO4 (5 μL) was stirred at room temperature for
12 h. The reaction mixture was extracted with EtOAc, washed
5-Methoxy-6,7-(dipentyloxy)flavone (41). To a stirred
with 10% aqueous NaHSO4 solution and water, dried over
solution of 40 (33 mg, 0.08 mmol) in a mixture of MeOH (4
MgSO4, and then concentrated in vacuo. The residue was
mL) and THF (8 mL) was added TMSCHN2 (2 M in hexanes,
recrystallized from MeOH to give compound 25 (26 mg, 57%)
0.32 mL, 0.64 mmol). The reaction mixture was stirred at room
as a yellow powder. mp 263-265 °C; 1H NMR (DMSO-d6) δ
temperature for 24 h and evaporated. Flash chromatography
7.07 (s, 1H), 7.59 (m, 3H), 8.12 (m, 2H), 9.58, 10.92, 12.76 (s,
of the residue, eluting with CH2Cl2/acetone (20:1), gave
3H); MS (EI) m/z 350 [M + 2]+, 348 ([M]+, base), 270.
compound 41 (32.6 mg, 98%) as a white solid. mp 107-108 6-Ethoxy-5,7-dihydroxyflavone (32) and 6,7-Diethoxy-
°C; 1H NMR (CDCl3) δ 0.95, 0.97 (t, 6H, J ) 6.6 Hz), 1.39-
5-hydroxyflavone (33). A mixture of 1 (81 mg, 0.3 mmol),
1.54 (m, 8H), 1.81, 1.93 (quintet, 4H, J ) 6.6 Hz), 3.99 (s, 3H),
4.03, 4.10 (t, 4H, J ) 6.6 Hz), 6.78 (s, 1H), 6.81 (s, 1H), 7.53
acetone (25 mL) was refluxed with stirring for 18 h. The
(m, 3H), 7.90 (m, 2H); MS (EI) m/z 424 [M]+.
reaction mixture was concentrated under reduced pressure,
6,7-(Dihexyloxy)-5-hydroxyflavone (42). A mixture of 1
diluted with water (50 mL), and extracted with CH2Cl2 (50
(54 mg, 0.2 mmol), 1-bromohexane (0.084 mL), and anhydrous
mL × 3). The extract was washed with water and dried over
K2CO3 (110 mg) in acetone (20 mL) was refluxed with stirring
MgSO4 and the solvent evaporated in vacuo. The residue was
for 24 h. The reaction mixture was concentrated under reduced
purified by flash chromatography on a column of silica gel and
pressure, diluted with water (40 mL) and extracted with CH2-
eluted with CH2Cl2/MeOH (50:1) to give compounds 32 (30 mg,
Cl2 (40 mL × 3). The extract was washed with water and dried
Anti-P-glycoprotein Activity of BaicaleinJournal of Medicinal Chemistry, 2004, Vol. 47, No. 22
over MgSO4 and the solvent evaporated in vacuo. The residue
were transferred to scintillation vials and counted with a
was purified by flash chromatography on a column of silica
-counter (Beckman, model LS 5000) after adding 10 mL
gel and eluted with CH2Cl2/MeOH (60:1) to give compound 42
scintillation fluids (SafeScint, American Bioanalytical Co.,
(71.4 mg, 82%) as a pale yellow powder. mp 96-97 °C; 1H NMR
Natick, MA). Data presented are the mean of three indepen-
(CDCl3) δ 0.92, 0.93 (t, 6H, J ) 6.6 Hz), 1.32-1.41 (m, 8H),
1.48-1.53 (m, 4H), 1.80, 1.90 (quintet, 4H, J ) 6.6 Hz), 4.05,
Growth Inhibitory Assay. Approximately 104 KB or KB/
4.09 (t, 4H, J ) 6.6 Hz), 6.56 (s, 1H), 6.68 (s, 1H), 7.54 (m,
MDR cells were seeded into 24-well tissue culture plates in
3H), 7.90 (m, 2H); MS (EI) m/z 438 [M]+.
RPMI 1640 medium plus 10% fetal bovine serum for 24 h, after
6,7-(Dihexyloxy)-5-methoxyflavone (43). To a stirred
which the cells were treated with various concentrations of
solution of 42 (47 mg, 0.1 mmol) in a mixture of MeOH (4 mL)
synthetic flavones in culture medium for 3 days. The cells were
then fixed and stained with methylene blue in 50% MeOH,
mL, 0.8 mmol). The reaction mixture was stirred at room
washed thoroughly with tap water, and dissolved with 0.5 mL
temperature for 24 h and evaporated. Flash chromatography
of 0.5% sarcosyl.39 The amount of cellular protein, which is
proportional to the cell number, is estimated by the absorption
compound 43 (41 mg, 91%) as a white solid. mp 93-95 °C; 1H
(OD595 nm). The growth inhibitory assay, which is presented
3) δ 0.92, 0.94 (t, 6H, J ) 6.6 Hz), 1.30-1.41 (m,
50 value, represents the concentration of compound
8H), 1.51-1.55 (m, 4H), 1.80, 1.92 (quintet, 4H, J ) 6.6 Hz),
required to inhibit 50% of cell growth. The cell doubling time
3.99 (s, 3H), 4.03, 4.10 (t, 4H, J ) 6.6 Hz), 6.76 (s, 1H), 6.81
of KB and KB/MDR cells is about 20 to 24 h. The data
(s, 1H), 7.52 (m, 3H), 7.90 (m, 2H); MS (EI) m/z 452 [M]+.
presented are the mean of three independent experiments. 6,7-Diethoxy-5-methoxyflavone (44). To a stirred solu- Acknowledgment. The corresponding author is a
tion of 33 (33 mg, 0.1 mmol) in a mixture of MeOH (4 mL) and THF (8 mL) was added TMSCHN
fellow of the National Foundation for Cancer Research.
mL, 0.8 mmol). The reaction mixture was stirred at room
Part of this work was supported by National Foundation
temperature for 24 h and evaporated. Flash chromatography
for Cancer Research (YCC) and the National Institutes
of the residue, eluting with CH2Cl2/acetone (10:1 to 5:1),
afforded compound 44 (33.8 mg, 99%) as a white solid. mp 126-128 °C; 1H NMR (CDCl3) δ 1.42, 1.55 (t, 6H, J ) 6.9 Hz), Supporting Information Available: HPLC analytical
4.00 (s, 3H), 4.13, 4.19 (q, 4H, J ) 6.9 Hz), 6.77 (s, 1H), 6.81
data. This material is available free of charge via the Internet
(s, 1H), 7.52 (m, 3H), 7.90 (m, 2H); 13C NMR (CDCl3) δ 14.94,
16.05, 62.39, 65.13, 70.22, 97.15, 108.78, 113.18, 126.37,129.36, 131.61, 132.10, 139.93, 153.27, 154.92, 157.94, 161.46,
References
177.67; MS (EI) m/z 340 [M]+. HRMS for C20H20O5 [M]+:
(1) Germann, U. A. P-glycoproteinsa mediator of multidrug resis-
calculated, 340.1311; found, 340.1315.
tance in tumour cells. Eur. J. Cancer 1996, 32A, 927-944. 6,7-Dibutoxy-5-hydroxyflavone (45). A mixture of 1 (81
(2) Gottesman, M. M.; Pastan, I. Biochemistry of multidrug resis-
mg, 0.3 mmol), 1-iodobutane (0.1 mL), and anhydrous K
tance mediated by the multidrug transporter. Annu. Rev.
(165 mg) in acetone (20 mL) was refluxed with stirring for 24
Biochem. 1993, 62, 385-427.
(3) Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M. M.; Pastan,
h. The reaction mixture was concentrated under reduced
I. et al. Cellular localization of the multidrug-resistance gene
pressure, diluted with water (50 mL), and extracted with CH2-
product P-glycoprotein in normal human tissues. Proc. Natl.
Cl2 (3 × 50 mL). The extract was washed with water and dried
Acad. Sci. U.S.A. 1987, 84, 7735-7738.
(4) Borst, P.; Schinkel, A. H. What have we learnt thus far from
4, and the solvent was evaporated in vacuo. The
residue was purified by flash chromatography on a column of
mice with disrupted P-glycoprotein genes? Eur. J. Cancer 1996, 32A, 985-990.
silica gel eluted with CH2Cl2/MeOH (60:1) to give compound
(5) Luker, G. D.; Nilsson, K. R.; Covey, D. F.; Piwnica-Worms, D. 45 (86 mg, 75%) as a yellow powder. mp 116-117 °C; 1H NMR
Multidrug resistance (MDR1) P-glycoprotein enhances esterifi-
(CDCl3) δ 0.99, 1.02 (t, 6H, J ) 6.6 Hz), 1.53-1.58 (m, 4H),
cation of plasma membrane cholesterol. J. Biol. Chem. 1999, 274,
1.79, 1.89 (quintet, 4H, J ) 6.6 Hz), 4.06, 4.10 (t, 4H, J ) 6.6
Hz), 6.56 (s, 1H), 6.68 (s, 1H), 7.54 (m, 3H), 7.90 (m, 2H); 13C
(6) Dalton, W. S.; Grogan, T. M.; Meltzer, P. S.; Scheper, R. J.; Durie,
B. G. et al. Drug-resistance in multiple myeloma and non-
3) δ 14.17, 14.31, 19.55, 19.62, 31.40, 32.61, 69.26,
Hodgkin’s lymphoma: detection of P-glycoprotein and potential
73.51, 91.65, 106.00, 106.52, 126.63, 129.47, 131.86, 132.13,
circumvention by addition of verapamil to chemotherapy. J. Clin.
132.49, 153.63, 153.71, 159.25, 164.17, 183.11; MS (EI) m/zOncol. 1989, 7, 415-424.
382 [M]+. HRMS for C23H26O5 [M]+: calculated, 382.1780;
(7) Miller, T. P.; Grogan, T. M.; Dalton, W. S.; Spier, C. M.; Scheper,
R. J. et al. P-glycoprotein expression in malignant lymphomaand reversal of clinical drug resistance with chemotherapy plus
6,7-Dibutoxy-5-methoxyflavone (46). To a stirred solu-
high-dose verapamil. J. Clin. Oncol. 1991, 9, 17-24.
tion of 45 (49 mg, 0.128 mmol) in a mixture of MeOH (5 mL)
(8) Nuessler, V.; Scheulen, M. E.; Oberneder, R.; Kriegmair, M.;
and THF (10 mL) was added TMSCHN2 (2 M in hexanes, 0.5
Goebel, K. J. et al. Phase I and pharmacokinetic study of the
mL, 1 mmol). The reaction mixture was stirred at room
P-glycoprotein modulator dexniguldipine-HCL. Eur. J. Med. Res.
temperature for 24 h and then evaporated. Flash chromatog-
1997, 2, 55-61.
(9) Wilson, W. H.; Jamis-Dow, C.; Bryant, G.; Balis, F. M.; Klecker,
raphy of the residue, eluting with CH2Cl2/acetone (15:1), gave
R. W. et al. Phase I and pharmacokinetic study of the multidrug
compound 46 (46 mg, 91%) as a white solid. mp 103-105 °C;
resistance modulator dexverapamil with EPOCH chemotherapy.
1H NMR (CDCl3) δ 0.99, 1.03 (t, 6H, J ) 6.6 Hz), 1.55-1.59
J. Clin. Oncol. 1995, 13, 1985-1994.
(m, 4H), 1.78, 1.91 (quintet, 4H, J ) 6.6 Hz), 3.99 (s, 3H), 4.04,
(10) Hyafil, F.; Vergely, C.; Du Vignaud, P.; Grand-Perret, T. In vitro
4.11 (t, 4H, J ) 6.6 Hz), 6.81 (s, 1H), 6.82 (s, 1H), 7.53 (m,
and in vivo reversal of multidrug resistance by GF120918, an
acridonecarboxamide derivative. Cancer Res. 1993, 53, 4595-
19.63, 31.37, 32.69, 62.44, 69.19, 74.33, 97.17, 108.78, 113.18,
(11) Boesch, D.; Gaveriaux, C.; Jachez, B.; Pourtier-Manzanedo, A.;
126.36, 129.34, 129.63, 130.21, 131.58, 132.13, 140.25, 153.18,
Bollinger, P. et al. In vivo circumvention of P-glycoprotein-
154.89, 158.11, 161.41, 177.68; MS (EI) m/z 396 [M]+. HRMS
mediated multidrug resistance of tumor cells with SDZ PSC 833. Cancer Res. 1991, 51, 4226-4233.
calculated, 396.1937; found, 396.1939.
(12) Ramu, A.; Spanier, R.; Rahamimoff, H.; Fuks, Z. Restoration of
Vinblastine Uptake by the KB/MDR Cells. KB/MDR
doxorubicin responsiveness in doxorubicin-resistant P388 mu-
cells,38 which overexpress human P-gp 170 protein, were
rine leukaemia cells. Br. J. Cancer 1984, 50, 501-507.
seeded into 24-well tissue culture plates in RPMI 1640 medium
(13) Middleton, E., Jr.; Kandaswami, C.; Theoharides, T. C. The
plus 10% fetal bovine serum for 24 h without the selecting
effects of plant flavonoids on mammalian cells: implications for
agent doxorubicin (37 nM). The cells were then treated with
inflammation, heart disease, and cancer. Pharmacol. Rev. 2000, 52, 673-751.
[3H]vinblastine plus synthetic flavones in HBSS for 60 min.
(14) Bailey, D. G.; Malcolm, J.; Arnold, O.; Spence, J. D. Grapefruit
The cells were washed twice with ice-cold PBS, harvested with
juice-drug interactions. Br. J. Clin. Pharmacol. 1998, 46, 101-
1 N NaOH, and neutralized with 1 N HCl. The cell aliquots
Journal of Medicinal Chemistry, 2004, Vol. 47, No. 22
(15) Ducharme, M. P.; Warbasse, L. H.; Edwards, D. J. Disposition
(27) Ferriola, P. C.; Cody, V.; Middleton, E., Jr. Protein kinase C
of intravenous and oral cyclosporine after administration with
inhibition by plant flavonoids. Kinetic mechanisms and structure-
grapefruit juice. Clin. Pharmacol. Ther. 1995, 57, 485-491.
activity relationships. Biochem. Pharmacol. 1989, 38, 1617-
(16) De Vincenzo, R.; Scambia, G.; Benedetti Panici, P.; Ranelletti,
F. O.; Bonanno, G. et al. Effect of synthetic and naturally
(28) Hagiwara, M.; Inoue, S.; Tanaka, T.; Nunoki, K.; Ito, M. et al.
occurring chalcones on ovarian cancer cell growth: structure-
Differential effects of flavonoids as inhibitors of tyrosine protein
activity relationships. Anticancer Drug Des. 1995, 10, 481-490.
kinases and serine/threonine protein kinases. Biochem. Phar-
(17) Murakami, S.; Muramatsu, M.; Tomisawa, K. Inhibition of
macol. 1988, 37, 2987-2992.
gastric H+, K(+)-ATPase by flavonoids: a structure-activity
(29) Perez-Victoria, J. M.; Chiquero, M. J.; Conseil, G.; Dayan, G.;
study. J. Enzyme Inhib. 1999, 14, 151-166.
Di Pietro, A. et al. Correlation between the affinity of flavonoids
(18) Akiyama, T.; Ishida, J.; Nakagawa, S.; Ogawara, H.; Watanabe,
binding to the cytosolic site of Leishmania tropica multidrug
S. et al. Genistein, a specific inhibitor of tyrosine-specific protein
transporter and their efficiency to revert parasite resistance to
kinases. J. Biol. Chem. 1987, 262, 5592-5595.
daunomycin. Biochemistry 1999, 38, 1736-1743.
(19) De Azevedo, W. F., Jr.; Mueller-Dieckmann, H. J.; Schulze-
(30) Lo, A.; Burckart, G. J. P-glycoprotein and drug therapy in organ
Gahmen, U.; Worland, P. J.; Sausville, E. et al. Structural basis
transplantation. J. Clin. Pharmacol. 1999, 39, 995-1005.
for specificity and potency of a flavonoid inhibitor of human
(31) Wang, R. B.; Kuo, C. L.; Lien, L. L.; Lien, E. J. Structure-activity
CDK2, a cell cycle kinase. Proc. Natl. Acad. Sci. U.S.A. 1996,
relationship: analyses of p-glycoprotein substrates and inhibi-
tors. J. Clin. Pharm. Ther. 2003, 28, 203-228.
(20) Boumendjel, A.; Di Pietro, A.; Dumontet, C.; Barron, D. Recent
(32) Di Pietro, A.; Conseil, G.; Perez-Victoria, J. M.; Dayan, G.;
advances in the discovery of flavonoids and analogues with high-
Baubichon-Cortay, H. et al. Modulation by flavonoids of cell
affinity binding to P-glycoprotein responsible for cancer cell
multidrug resistance mediated by P-glycoprotein and related
multidrug resistance. Med. Res. Rev. 2002, 22, 512-529.
ABC transporters. Cell Mol. Life Sci. 2002, 59, 307-322.
(21) Efferth, T.; Davey, M.; Olbrich, A.; Rucker, G.; Gebhart, E. et
(33) Chambers, T. C.; Pohl, J.; Raynor, R. L.; Kuo, J. F. Identification
al. Activity of drugs from traditional Chinese medicine toward
of specific sites in human P-glycoprotein phosphorylated by
sensitive and MDR1- or MRP1-overexpressing multidrug-
protein kinase C. J. Biol. Chem. 1993, 268, 4592-4595.
resistant human CCRF-CEM leukemia cells. Blood Cells Mol.
(34) Callaghan, R.; Higgins, C. F. Interaction of tamoxifen with the
Dis. 2002, 28, 160-168.
multidrug resistance P-glycoprotein. Br. J. Cancer 1995, 71,
(22) Conseil, G.; Baubichon-Cortay, H.; Dayan, G.; Jault, J. M.;
Barron, D. et al. Flavonoids: a class of modulators with
(35) Smith, C. D.; Zilfou, J. T. Circumvention of P-glycoprotein-
bifunctional interactions at vicinal ATP- and steroid-binding
mediated multiple drug resistance by phosphorylation modula-
sites on mouse P-glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 1998,
tors is independent of protein kinases. J. Biol. Chem. 1995, 270,
(23) Perez-Victoria, J. M.; Perez-Victoria, F. J.; Conseil, G.; Maitre-
(36) Conseil, G.; Perez-Victoria, J. M.; Jault, J. M.; Gamarro, F.;
jean, M.; Comte, G. et al. High-affinity binding of silybin
Goffeau, A. et al. Protein kinase C effectors bind to multidrug
derivatives to the nucleotide-binding domain of a Leishmania
ABC transporters and inhibit their activity. Biochemistry 2001,
tropica P-glycoprotein-like transporter and chemosensitization
of a multidrug-resistant parasite to daunomycin. Antimicrob
(37) Dayan, G.; Jault, J. M.; Baubichon-Cortay, H.; Baggetto,
Agents Chemother 2001, 45, 439-446.
L. G.; Renoir, J. M. et al. Binding of steroid modulators to
(24) Thiyagarajah, P.; Kuttan, S. C.; Lim, S. C.; Teo, T. S.; Das, N.
recombinant cytosolic domain from mouse P-glycoprotein in close
P. Effect of myricetin and other flavonoids on the liver plasma
proximity to the ATP site. Biochemistry 1997, 36, 15208-15215.
membrane Ca2+ pump. Kinetics and structure-function rela-
(38) Chen, H. X.; Bamberger, U.; Heckel, A.; Guo, X.; Cheng, Y. C.
tionships. Biochem. Pharmacol. 1991, 41, 669-675.
BIBW 22, a dipyridamole analogue, acts as a bifunctional
(25) Hirano, T.; Oka, K.; Akiba, M. Effects of synthetic and naturally
modulator on tumor cells by influencing both P-glycoprotein and
occurring flavonoids on Na+, K+-ATPase: aspects of the
nucleoside transport. Cancer Res. 1993, 53, 1974-1977.
structure-activity relationship and action mechanism. Life Sci.
(39) Park, S. Y.; Lam, W.; Cheng, Y. C. X-ray repair cross-
1989, 45, 1111-1117.
complementing gene I protein plays an important role in
(26) Jinsart, W.; Ternai, B.; Polya, G. M. Inhibition of rat liver cyclic
camptothecin resistance. Cancer Res. 2002, 62, 459-465.
AMP-dependent protein kinase by flavonoids. Biol. Chem. Hoppe Seyler 1992, 373, 205-211.
ProStrakan Group plc ProStrakan Announces FDA Acceptance of US Submission of Cellegesic™ (nitroglycerin) 0.4% Ointment Galashiels, UK. 21st October 2009 – ProStrakan Group plc (LSE: PSK), the international specialty pharmaceutical company, today announces that the US Food and Drug Administration (“FDA”) has confirmed acceptance for review of ProStrakan’s refiling of its
Email: [email protected]—Recently, several research papers in the area ofcertainly are not as fundamental to humankind such as, forinformation security were published that may or may not beexample, stem cell research or other issues in natural science,considered unethical. Looking at these borderline cases is relevantwe still feel the need to address these ethica
 Anti-P-glycoprotein Activity of Baicalein
Journal of Medicinal Chemistry, 2004, Vol. 47, No. 22
Scheme 1a
Anti-P-glycoprotein Activity of Baicalein
Journal of Medicinal Chemistry, 2004, Vol. 47, No. 22
Scheme 1a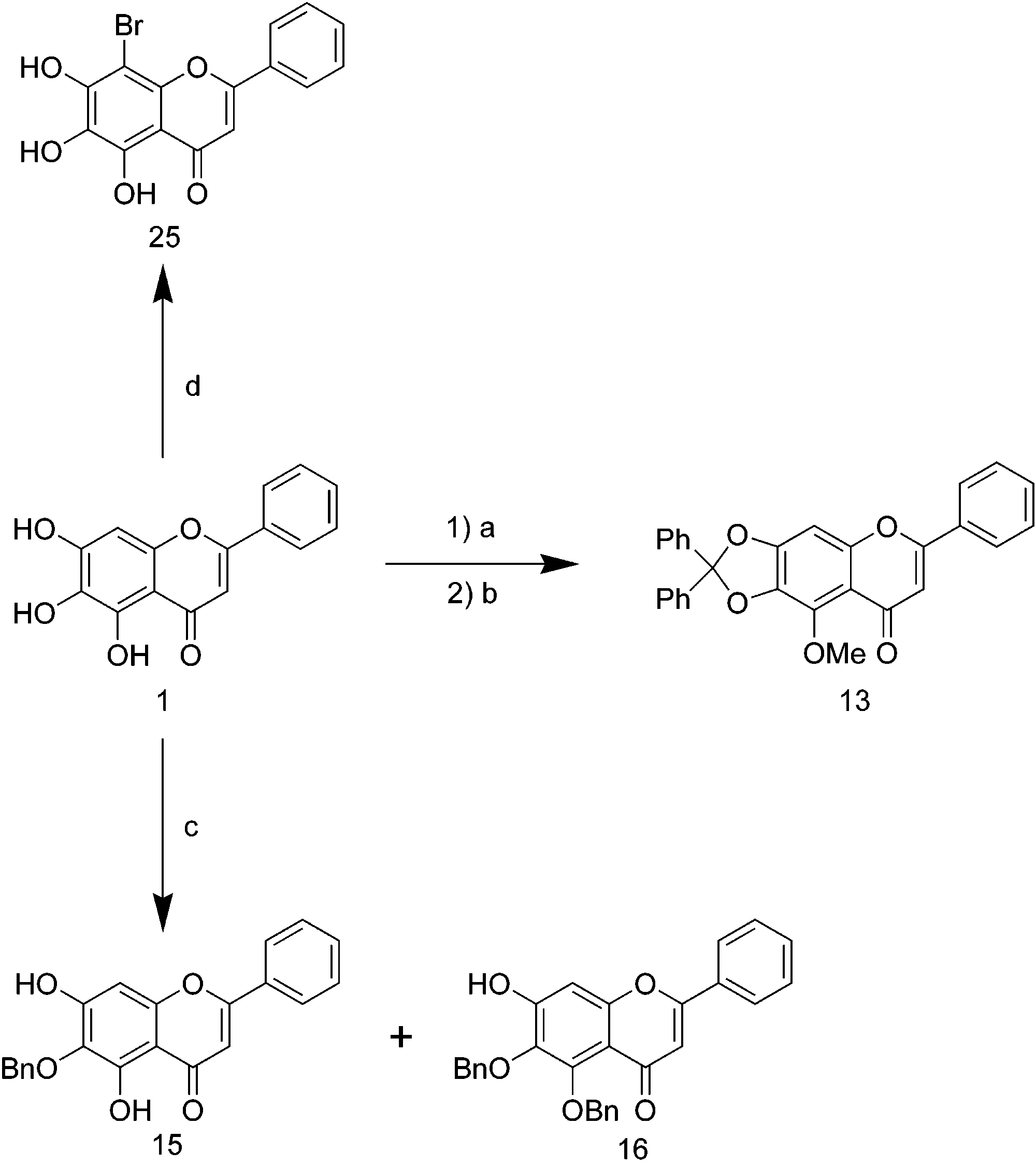 Journal of Medicinal Chemistry, 2004, Vol. 47, No. 22
Scheme 3a
Journal of Medicinal Chemistry, 2004, Vol. 47, No. 22
Scheme 3a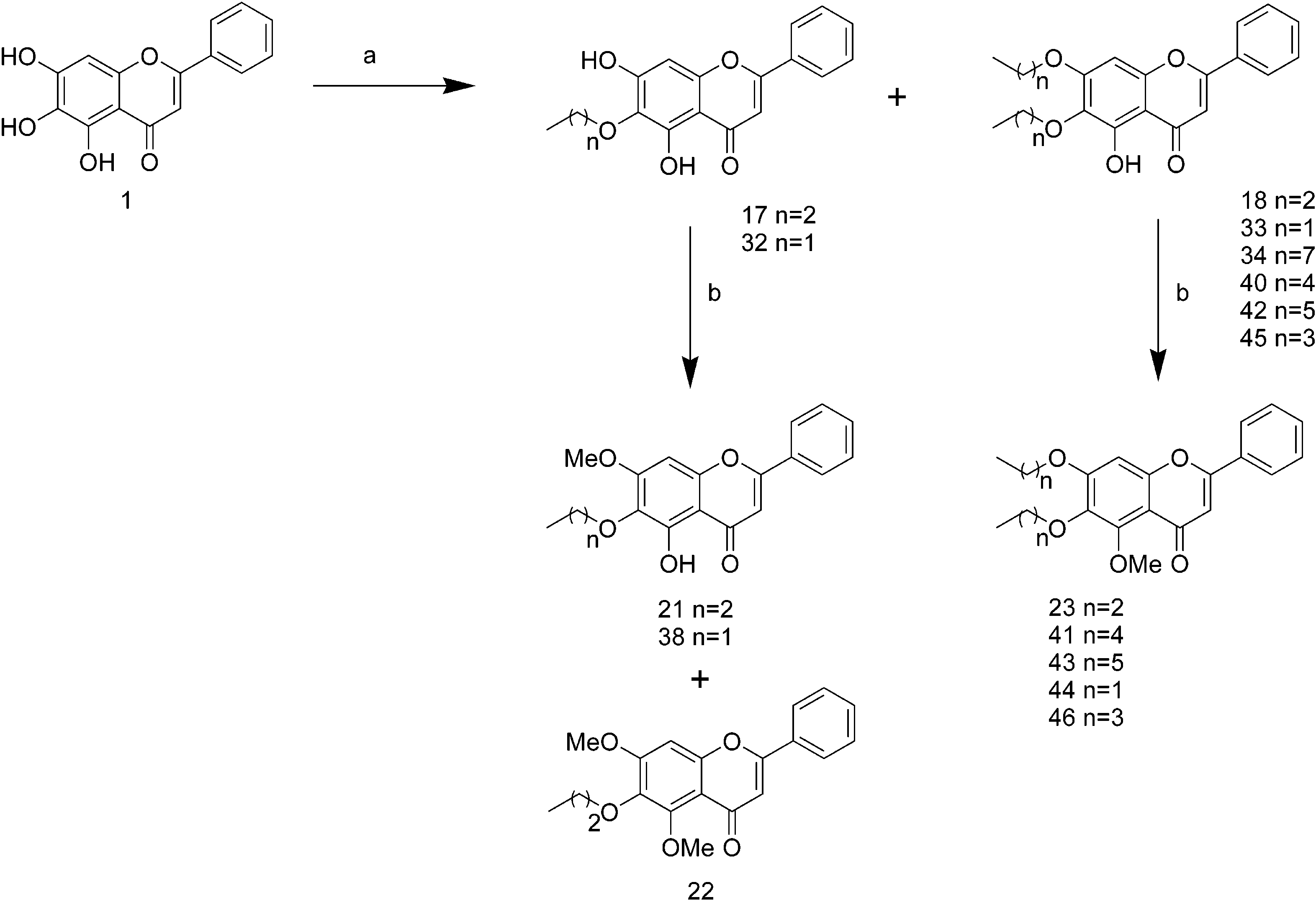 Anti-P-glycoprotein Activity of Baicalein
Journal of Medicinal Chemistry, 2004, Vol. 47, No. 22
Scheme 4a
Anti-P-glycoprotein Activity of Baicalein
Journal of Medicinal Chemistry, 2004, Vol. 47, No. 22
Scheme 4a